Katelyn McNair focuses on Computational Biology, applying mathematical and computational methods to biological problem. McNair mainly focuses on the fields of bacterial and viral genomics, but has worked on many diverse projects: from ecology to image recognition.
Research Advisor(s)
Anca Segall
Research Abstract
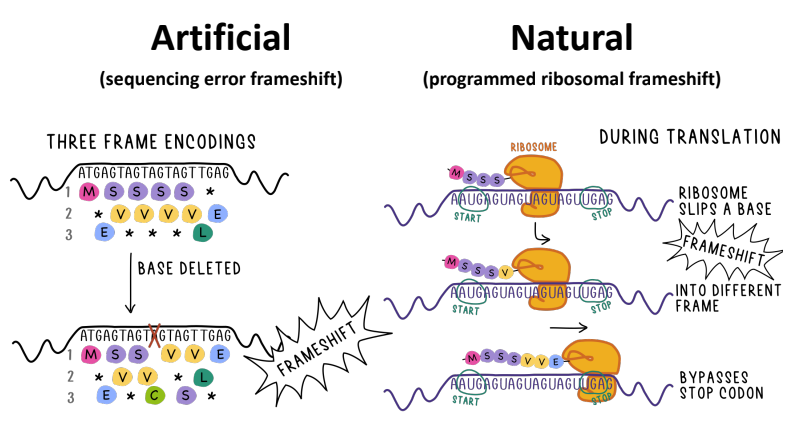
Genomic sequencing is at the forefront of biological research, in part due to the ever-increasing accessibility of sequencing technology. What used to take an entire lab devoted solely to nucleotide sequencing, can now be accomplished by a single person on a tiny USB device plugged into an old laptop. Around 80% of this data is microbial in nature: originating from Bacteria, Archaea, or viruses. The goal of sequencing is to identify and analyze the genes that occur within the DNA (or RNA). This is because it is the genes that carry out the functions and are the building blocks of every cell. Frameshifts, where coding sequences switch between frames on the collinear strand, are ubiquitous in this genomic data, as both artificially induced sequencing error and naturally occurring cellular processes. These frameshifts break the genes into different frames, which confound downstream gene prediction analysis; since all current genome-based analysis of microbes uses methods that are remiss when it comes to identifying these frameshifts. In order to improve current genomic research, we developed software that is able to identify both artificial and natural frameshifts within an input genome, using amino-acid translations of a scrolling windows through the nucleotides, and an artificial neural network to identify those windows that come from a coding region. Then a change-point algorithm is used to identify when a coding frameshift occurs in the absence of start and stop codons.